Do Methylation Changes Drive the Adaptation to Altered Light Environment in Guppies?
2021-11-22
Chapter 1 Introduction
1.1 Literature review
1.1.1 Experimental evolution
Experimental evolution is the study of evolutionary processes in isolated populations through experiments which involve the manipulation of a population’s environment. The purpose of this type of experimentation is to explore evolutionary dynamics. This exploration is undertaken through investigating adaptations and how they arise, estimating evolutionary parameters and testing evolutionary hypotheses (Kawecki et al. 2012). While evolution is commonly thought of as a process that is only able to be seen when observed from a distance in time, in reality it is an ongoing process in all species. By creating experimental scenarios and examining the effects of these conditions on different populations we have the potential to gain insights into the different mechanisms and dynamics governing evolution. This research leads to a greater understanding of the fundamental principles of evolution (Lenski 2017).
Experiments of this kind most frequently study the shifting of alleles within a population that confer an evolutionary advantage within a particular environment. However, many experiments which are on a longer time scale and/or focus on organisms that have a short generation time also study the effects of mutations on evolution. The two essential mechanisms governing evolution, gene flow and genetic drift, are also incorporated into some experimental evolution studies (Kawecki et al. 2012).
Experimental evolution research can often be conducted in ways which are unique to the field of study. For one, experimental evolution experiments are frequently done on far longer time scales than most other forms of research. Most scientific experiments set up a scientific question, control for the variables which could impede their answering of the question, and then proceed with experimentation for a predetermined time or until their question is answered. While these standard experiments are common in the field, much of experimental evolution does not proceed this way. Instead, experiments may be carried out indefinitely. While the ongoing experiment is proceeding, variables may be altered and the populations involved may be examined and tested to answer scientific questions. For example, Lenski’s experiments involving E. coli have been ongoing from the original founding population established in 1988 till this very day (Barrick et al. 2009).
Another way in which this type of research differs from regular experiments is that the scientific questions being answered are not necessarily known to the experimenter until after the examination and testing of the populations has begun. The hypothesis devised before the experimental evolution research began may initially be narrow. However, the nature of creating a system and allowing itself to run its course ultimately reveals many phenotypic and genotypic adaptations to that environment that were not initially conceived (Kawecki et al. 2012).
The study this project is based upon is an experiment which involves breeding multiple populations of guppies under altered light conditions. This experiment is of the indefinite kind and therefore has and will continue to lead to new scientific questions that can be answered through different examinations and tests (Kranz et al. 2018).
Lenski’s E coli experiment mentioned earlier is another example of this indefinite kind. The isolation of multiple populations of E. coli from a single ancestral colony was originally devised to test mean fitness selection but has gone on to be used to study other evolutionary concepts such as correlated responses, pleiotropy, historical contingency, evolvability, epistasis, specialisation, forces maintaining diversity, the origin of a new function (Kawecki et al. 2012), genome evolution (Barrick et al. 2009), convergent/divergent evolution (Lenski 2017), and the evolution of high mutation rates (Sniegowski et al. 1997).
1.1.2 Guppies as a model of sexual selection
Guppies are an excellent model of sexual selection for many reasons. They are internally fertilizing livebearers that are easy to maintain in aquaria and have a short generation time. Both males and females are selective when choosing their mate and this selection is not based on resources (Auld et al. 2016). Their sexual selective system system is a female-based polygyny (Kodric-Brown 1985). Guppies are a sexually dimorphic species where males are typically smaller and more colourful than their female counterparts (Auld et al. 2016).
The competition among males for reproductive success is great due to their natural population density. The intensity of courtship, composition of male group traits, sperm competition and the colour patterns on the surface of the males are the essential factors governing male success (Kodric-Brown 1985). Females prefer males which are more brightly coloured and larger in size (Auld et al. 2016).
Because guppies breed through internal fertilisation they are capable of making more specific choices in regard to their mating. This trait also means that the females are multiply mated and therefore sperm competition plays an essential role in reproductive success (Auld et al. 2016). Even though sperm competition is a physiological determinant of reproductive success, the presence of this physiological factor has an impact on the behaviour of the guppies. For example, Poecilia reticulata males have demonstrated the behaviour of altering their selection for their ideal mate in response to the presence of larger males. (Auld et al. 2016).
Guppies are an ideal model for visual and colouration governed sexual selection. Firstly, they are a premier model organism for phenotypic variation as they adapt rapidly in response to sexual and predation selection changes (Houde 1997). Secondly, their opsin gene expression has been demonstrated to be very plastic (Kranz et al. 2018). Lastly, the surface colouration of guppies is highly heritable, genetically polymorphic and plays an important role in sexual selection. The pattern, size and colouration of their surface prints are extremely complex and varied (Kranz et al. 2018).
1.1.3 The use of different light environments to simulate environmental changes and as a tool to study sexual selection
There is a research vacuum regarding how the light conditions of an environment impacts natural or sexual selection. Studies related to this field have previously involved studying the effects of the natural environment rather than simulating environmental conditions (Kranz et al. 2018).
An example of this includes a study on the opsin gene expression in birds of the same family Parulidae. Individuals from populations and species which had died due to their collision with buildings were collected and studied. It was found that the opsin gene expression differed significantly between both the species and populations. The different populations evolved in separate environments and therefore had varied light conditions which resulted in plumage dichromatism. The researchers concluded that light conditions altered opsin gene expression through natural and sexual selection (Bloch 2015).
A rare example of a simulation of altered light environments to observe their effect on sexual selection was done on three-spined sticklebacks, Gasterosteus aculeatus. This was an experimental study on the effect of increased filamentous algae in the environment and its cost on mating for individuals. While this experiment was designed to understand the effects of altered light conditions on sexual selection, it differed significantly from the experiment on which this project is based as it did not track phenotypic changes over many generations (Candolin et al. 2007).
The experiment this project is based upon did not only simulate altered light environment conditions as a tool to understand its role in sexual selection but also tracked the effects of these conditions over multiple generations (~20). The results revealed that light conditions are a primary factor in determining the physiology of guppies through sexual selection (Kranz et al. 2018).
A study derived on data obtained from this same experiment focused on the alteration of opsin expression. Vision is a practical trait to focus on when studying experimental evolution as light conditions are ever changing across all time scales and it is essential for organisms to adapt to these conditions in order to find sustenance and a mate. Because of this, the visual system has likely evolved to be adaptable in many organisms. As expected, opsin genes were highly plastic and were tuned to the particular light condition in which the guppies were bred (Kranz et al. 2018).
Another study was performed using this experimental system which focused on male colour pattern evolution. The effectiveness of colour pattern signal efficiency for sexual selection under altered light conditions was observed. Divergent light conditions lead to diverged conspicuousness of males to females. This conspicuousness came from a match of hue, chroma and luminance to the simulated environmental light. This type of experimentation which occurred over many generations to test opsin gene expression and male colouration pattern evolution was a first of its kind (Kranz et al. 2018).
1.1.4 The role of genotype on phenotype
The central dogma of molecular biology, as it is most commonly thought of, describes the pathway from the genotype of an organism to its phenotype. This process of DNA to RNA to protein is depicted in the following diagram.
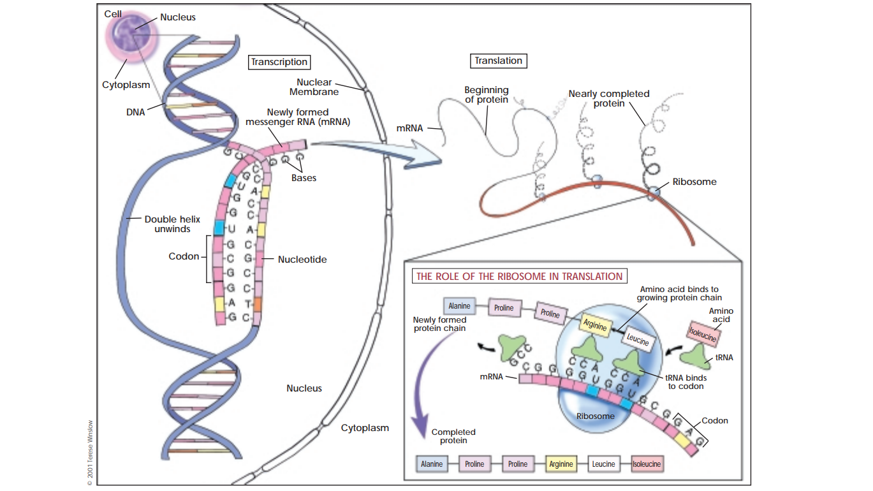
Figure 1.1: Figure 1 - Diagram depicting Transcription, translation, and protein synthesis (Department of Health and Human Services 2001)
The original conception of the central dogma of molecular biology developed by Francis Crick in 1957 differs significantly from the modern conception of the term. Today, the central dogma of molecular biology is thought of as the basic but fundamental idea that DNA is the template for RNA which is sequentially the template for proteins. However, Francis Crick’s central dogma of molecular biology was actually a series of hypotheses which were widely held by most in the field (Crick 1970). This original central dogma most importantly focused on information transfer, particularly that once information is transferred to proteins it does not leave the proteins to be transferred elsewhere. The original conception of the term has stood the test of time and has remained mostly accurate (Strasser 2006).
As our understanding of molecular biology has developed, the complexity of information transfer and the effect of genotype on phenotype has slowly been uncovered. There are now many challenges to the simple pathways of transcription to translation to protein which many people still believe to be the central dogma of molecular biology. Reverse transcriptase allows RNA to be transferred back to DNA. Prions change the structure of other proteins. Chaperones are proteins which play an important role in the process of protein synthesis. RNA can be spliced into alternate forms of mRNA which are used to create different proteins. Nucleotides can be added to RNA so that the final mRNA is not identical to its original DNA template. Finally, the expression of genes can be altered via many mechanisms (Crick 1970).
1.1.5 The role of epigenotype on phenotype
While the genotype is the primary determinant of the phenotype, this understanding of genetics only observes a part of the full picture. The expression of genes, that is the activation and translation of genes, plays an essential role in determining the phenotype. This expression of genes is regulated by DNA methylation, chromatin alterations (histone modifications and chromatin remodelling), miRNAs and interfering micro RNAs (Strasser 2006).
The term epigenetics was originally defined in the 1940s by Conrad Waddington. This original definition of the term was “the branch of biology which studies the casual interactions between genes and their products which bring the phenotype into being.”The original definition was designed to answer how a single common genome could generate many vastly different cell types within an organism (Sun et al. 2013). However, as our understanding of genetics has advanced the definition of the word has narrowed (Dupont et al. 2009). Today, the term could be defined as “potentially heritable, and environmentally-modifiable changes in gene expression that are mediated via non-DNA-encoded mechanisms” (Russo et al. 1996). Epigenetics is the mechanism by which environmental stimuli is translated into the regulation of gene function and expression in cell lineages (Sun et al. 2013).
DNA methylation is a stable epigenetic marker. It occurs when DNA methyltransferases attach a methyl group to a cytosine (Dunn and Smith 1958). A methyl group can be attached to adenine and act as an epigenetic marker in non-mammal organisms. The methyl group is added to the fifth position of the cytosine to form 5-methylcytosine (DNMTs) (Kim and Costello 2017).
DNA methylation plays a very important role in gene expression and mostly occurs on dinucleotide sites known as CpG sites. These sites are characterised by a cytosine followed by a guanine in the DNA sequence. Approximately 60-80% of CpG sites are methylated in humans and 7% of all CpG sites are located in regions known as CpG islands which are extremely CpG dense (Kim and Costello 2017). CpG islands make up approximately 60% of human gene promoter regions (Virani et al. 2012). In CpG dense promoter regions, DNA methylation is strongly linked to gene repression whereas in less CpG dense regions of the genome and within the gene body the DNA methylation can either activate or repress gene activity (Bock 2012). The repression of gene activity occurs in promoter regions as the DNA methylation can recruit proteins which are involved in gene repression or physically block the binding of transcription factors to particular sites (Moore et al. 2013).
Histones are the primary DNA packaging proteins around which DNA winds to form complexes called nucleosomes. A nucleosome is the basic unit of chromatin. Nucleosome organisation determines DNA transcription and is therefore an epigenetic factor. Nucleosome structure is categorised into euchromatin and heterochromatin (Virani et al. 2012).
Euchromatin is the more loosely packed state of the nucleosome which allows it to be transcriptionally active. Heterochromatin is far more compact and consequently is permanently silenced. However, facultative heterochromatin is silenced transcriptionally but has the potential to be unsilenced in response to specific environmental or genetic cues. This chromatin structure is regulated through chemical alterations with the primary alteration being covalent modifications to the N-terminus tails of histone proteins (Virani et al. 2012).
Two significant histone modifications include histone methylation and acetylation. Methylation on the lysine and arginine residues of histone have been linked to both the suppression and activation of transcription activity. Histone acetylation causes alterations in the chromatin structure which enable transcription. Lysine residue acetylation of the histones neutralises their charge which decreases their attraction to the negatively charged DNA and therefore opens up the structure of the molecule to be read (Virani et al. 2012).
microRNA (miRNA) has a significant effect on gene expression. They are short, approximately 22 nucleotides in length and are encoded in the genome. microRNA bind imperfectly to their target mRNA which may lead to the degradation or disruption of the mRNA’s capacity to be translated (Chuang and Jones 2007). miRNA have the potential to regulate epigenetics through regulating the expression of histone modifier molecules, meaning that they control chromatin configuration (Chuang and Jones 2007). Short interfering RNA (siRNA) are similar to miRNA in their nature and function, however siRNA differ in that they are double stranded and bind perfectly to their target mRNA (Caillaud et al. 2020).
1.1.6 Methods to perform genomic and methylation profiling
The first attempt to perform DNA sequencing occurred in 1968 when Ray Wu profiled 12 bases in the cohesive ends of bacteriophage lambda DNA (Wu and Kaiser 1968). Through the rest of the 1900s many methods were developed to sequence DNA and the process gradually became more efficient (Shendure et al. 2017). The development of revolutionary techniques included Sanger sequencing developed in 1977 (Sanger and Coulson 1975) and shotgun sequencing in 1979 (Staden 1979).
Techniques for sequencing of DNA continued to advance through the 90s and early 2000s and hierarchical shotgun sequencing was used to complete the human genome project in 2004. ‘Massive parallel’ or ‘next generation sequencing’ dramatically surpassed the efficiency of all other previously used techniques and is the standard for genomic profiling today (Shendure et al. 2017).
Whole genome bisulfite sequencing was first introduced in 1992 and has long been the ‘gold standard’ in methylation profiling (Hayatsu 2008). This technique involves treating DNA with bisulfite to convert unmethylated cytosine to uracil, leaving the methylated cytosines intact. When the genome is sequenced, the observable cytosines are only those which are methylated (Kurdyukov and Bullock 2016). However, bisulfite sequencing has its flaws such as its potential to damage DNA and introduce bias to sequencing data. The NEBnext® enzymatic methyl-sequencing kit (EM-seq kit) was developed to overcome these problems. This kit functions similarly to bisulfite sequencing but it uses a series of enzymes to convert the unmethylated cytosines to uracil rather than using bisulfite as a chemical treatment (Vaisvila et al. 2019).
Other techniques to perform whole genome methylation profiling include HPLC-UV, LC-MS/MS, ELISA-Based methods, LINE-1 + Pyrosequencing, AFLP/RFLP, and LUMA (Kurdyukov and Bullock 2016). HPLC-UV is used to quantify the amount of deoxycytidine (dC) and methylated cytosines (5 mC) present in a hydrolysed DNA sample. The technique uses high performance liquid chromatography to determine the 5 mC/dC ratio for each sample (Kuo et al. 1980) Liquid chromatography can be combined with tandem mass spectrometry, which is referred to as LC-MS/MS, as an alternative to HPLC-UV that requires less quantity of DNA to be run (Song et al. 2005).
ELISA-Based methods involve capturing segments of the genome on an ELISA plate and using sequential incubation steps to detect methylated cytosines (Kurdyukov and Bullock 2016). LINE-1 + Pyrosequencing involves treating DNA with bisulfite before the PCR amplification of LINE-1 conservative sequences. The methylated cytosines are then detected using pyrosequencing (Ogino et al. 2008). Standard PCR-based amplification fragment length polymorphism (AFLP) and restriction fragment length polymorphism (RFLP) can both be utilised to perform whole genome methylation profiling. Both methods are cheap, however they are only capable of detecting a small percentage of the total DNA methylation (Kurdyukov and Bullock 2016). LUMA is a technique that simultaneously utilises restriction enzymes which only cleave DNA that have been methylated restriction enzymes which are insensitive to DNA methylation. Bioluminometric polymerase extension assays are then used to quantify the extent of restriction cleavage to determine the amount of unmethylated DNA present in the sample (Karimi et al. 2006).
1.2 Aims
Prior research (by Larry Croft) has found genetic polymorphisms which are fundamental in creating sexually selected phenotypic changes within multiple populations of P. reticulata. However, how exactly these genetic polymorphisms manifest adaptations within the populations remains undetermined. They may be produced through the altering of gene function or through creating epigenetic changes.
Primary aims of the project:
To determine if sexually selected adaptive phenotypic changes in vertebrate populations arise because of the alteration of gene expression due to DNA methylation.
If DNA methylation does contribute to phenotypic changes, to identify which genes these are.
To identify if any biological functions/pathways have been enriched.
The scientific question we aim to answer:
- Do methylation changes contribute to short scale evolution in vertebrates?
1.3 Methods to be used
The first step in addressing these aims will be to perform a series of differential methylation analysis to identify methylome divergences between a foundation population of P. Reticulata and populations which have gone through multiple generations under altered light conditions. Three different light conditions were created, each replicated 3 times, making 10 separate populations to be analysed.
Once the data has been gathered and processed using the NovaSeq system and a bioinformatics pipeline, the differential methylation analysis will be conducted using methylkit and dmrseq. The methylkit analysis will be performed at both the CpG site scale and region scale. This analysis will allow identification of CpG sites/regions that are significantly differentially methylated between light condition groups. These sites will then be annotated to genes and the promoter regions of genes to determine those that may have had their expression altered due to DNA methylation modifications. Genes which could contribute to the known phenotypic changes are likely to have been created by the experimental conditions as sexually selected adaptations.
If a large number of significant CpG sites and/or regions are identified in the differential methylation analysis then an enrichment analysis will be run to discern if any biological pathways/functions may have been altered due to DNA methylation changes.
1.4 Expected results
The null hypothesis for the primary aim of this research is that the observable phenotypic variation created by the experimental conditions are completely due to altered gene function rather than altered gene expression. I hypothesise that the results of this project will indicate that genetic variation has been created through a combination of both altered gene function and altered gene expression. That is to say that there will be identifiable DNA methylation modifications that contribute to the known phenotypic divergences between the populations. I also predict that biological pathways/functions that contribute to the known phenotypic divergences will be significantly enriched.
1.5 Significance of the project
This experiment is globally significant as it is a world’s first in the field. An experimental evolution study which focuses on the transcriptome and epigenetic states in vertebrates over many generations has not yet been performed and has the potential to be a first step in revealing ground-breaking scientific knowledge. The project has the potential to develop new perspectives and discover essential information regarding the molecular processes which govern Darwinian selection over long time scales.
The findings garnered from this study may also have value in industries which rely on the selective breeding of vertebrates. Industries which would fall into these categories include agriculture, aquaculture and the pet industry.